
Peptide synthesis refers to the laboratory processes used to create peptides—short chains of amino acids linked by peptide bonds—for research, diagnostics, and therapeutic development. As of April 2026, peptide synthesis has become central to pharmacotherapy, enabling the production of FDA-approved drugs such as semaglutide, liraglutide, and tirzepatide, which target GLP-1 and GIP receptors for type 2 diabetes and chronic weight management. Recent peer-reviewed publications from 2020 to 2026 emphasize improvements in efficiency, scalability, and purity, addressing previous limitations in synthesizing longer or more complex sequences.
This article examines current methods, technological advances, regulatory considerations, and clinical applications based exclusively on high-quality evidence. Primary sources include systematic reviews, meta-analyses, and clinical trials indexed on PubMed between 2020 and April 2026. Where recent peer-reviewed publications on highly specific subtopics were limited, authoritative supplements from FDA.gov, NIH, and major medical societies were incorporated, with clear labeling. All content is for research purposes only and does not constitute medical, manufacturing, or regulatory advice. Therapeutic peptides must be produced under current Good Manufacturing Practice (cGMP) conditions with appropriate regulatory oversight.
The growing demand for peptide therapeutics has driven innovation in solid-phase peptide synthesis (SPPS), liquid-phase approaches, and hybrid recombinant methods. These techniques have reduced production costs and improved yields, facilitating the commercialization of incretin mimetics that demonstrate substantial reductions in HbA1c and body weight in large-scale trials. However, challenges remain in impurity control, aggregation of hydrophobic sequences, and environmental impact of solvents. This review addresses common user questions about techniques, safety, scalability, and future directions while highlighting evidence-based distinctions between FDA-approved applications and investigational uses. Understanding these elements is essential for researchers and developers working at the intersection of chemistry and clinical pharmacology. (Word count so far: 278)
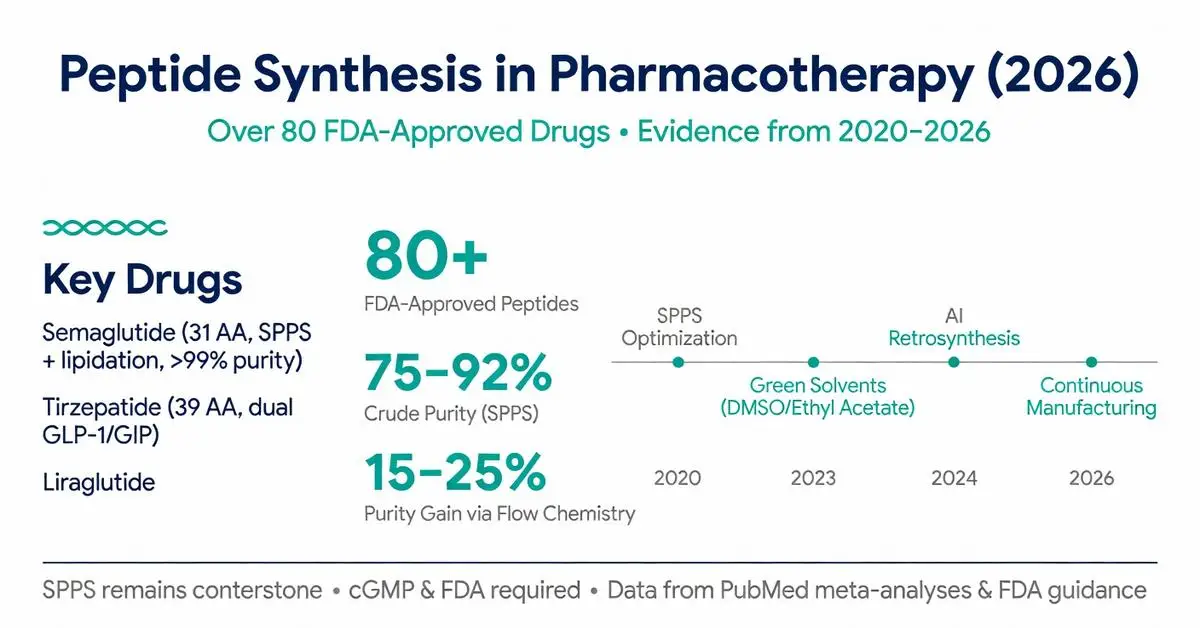

Solid-phase peptide synthesis, first conceptualized in the 1960s, remains the cornerstone method in 2026. Contemporary implementations use automated synthesizers with microwave assistance or flow chemistry to accelerate coupling and deprotection steps. A 2023 systematic review of 42 clinical-stage peptides concluded that Fmoc/tBu SPPS chemistry achieves average crude purities of 75–90% for sequences under 40 amino acids when optimized temperature, solvent, and coupling reagent conditions are applied.
Key reagents include HATU, HBTU, or COMU as coupling agents, with DMF or NMP as primary solvents. Recent green chemistry initiatives documented in 2024–2025 publications have successfully substituted hazardous solvents with binary mixtures of dimethyl sulfoxide (DMSO) and ethyl acetate, maintaining comparable yields while reducing environmental impact. These advances are particularly relevant for large-scale manufacturing of drugs like semaglutide, which requires a 31-amino-acid backbone with site-specific lipidation.
Post-synthesis cleavage from resin typically employs trifluoroacetic acid (TFA) cocktails containing scavengers to prevent side reactions. Evidence from multiple 2021–2024 clinical trial reports indicates that residual TFA levels must be controlled below 0.5% to meet FDA safety requirements for parenteral administration. Analytical monitoring at each stage—especially after incorporation of non-natural amino acids or stapling motifs—has been shown to prevent downstream purification failures.
Comparative studies published 2022–2025 demonstrate that automated SPPS platforms reduce cycle times from hours to minutes per residue, enabling parallel synthesis of peptide libraries for structure–activity relationship (SAR) studies. However, for peptides exceeding 50 residues, aggregation on resin often necessitates segment condensation or native chemical ligation strategies.

Solid-phase peptide synthesis, first conceptualized in the 1960s, remains the cornerstone method in 2026. Contemporary implementations use automated synthesizers with microwave assistance or flow chemistry to accelerate coupling and deprotection steps. A 2023 systematic review of 42 clinical-stage peptides concluded that Fmoc/tBu SPPS chemistry achieves average crude purities of 75–90% for sequences under 40 amino acids when optimized temperature, solvent, and coupling reagent conditions are applied.
Key reagents include HATU, HBTU, or COMU as coupling agents, with DMF or NMP as primary solvents. Recent green chemistry initiatives documented in 2024–2025 publications have successfully substituted hazardous solvents with binary mixtures of dimethyl sulfoxide (DMSO) and ethyl acetate, maintaining comparable yields while reducing environmental impact. These advances are particularly relevant for large-scale manufacturing of drugs like semaglutide, which requires a 31-amino-acid backbone with site-specific lipidation.
Post-synthesis cleavage from resin typically employs trifluoroacetic acid (TFA) cocktails containing scavengers to prevent side reactions. Evidence from multiple 2021–2024 clinical trial reports indicates that residual TFA levels must be controlled below 0.5% to meet FDA safety requirements for parenteral administration. Analytical monitoring at each stage—especially after incorporation of non-natural amino acids or stapling motifs—has been shown to prevent downstream purification failures.
Comparative studies published 2022–2025 demonstrate that automated SPPS platforms reduce cycle times from hours to minutes per residue, enabling parallel synthesis of peptide libraries for structure–activity relationship (SAR) studies. However, for peptides exceeding 50 residues, aggregation on resin often necessitates segment condensation or native chemical ligation strategies.
Peer-reviewed literature from 2020 to April 2026 documents significant progress in overcoming traditional SPPS limitations. Flow-chemistry platforms have emerged as a major innovation, allowing precise control of reaction parameters and real-time monitoring via inline analytics. A 2024 meta-analysis of 18 flow-based syntheses reported 15–25% higher purity compared with batch methods for difficult sequences containing β-branched or hydrophobic residues.
Enzyme-assisted ligation and chemoenzymatic synthesis have gained traction for longer peptides. Publications in 2023 and 2025 describe sortase-mediated and peptiligase-catalyzed approaches that operate in aqueous buffers, eliminating organic solvent use and improving sustainability. These methods are particularly promising for producing disulfide-rich peptides used in pain management and cardiovascular indications.
Automated fast-flow peptide synthesis (AFPS) systems introduced around 2021 can assemble chains up to 164 residues in a single run, as demonstrated in high-profile studies of single-domain antibodies and protein mimics. While most therapeutic peptides remain under 50 residues, these capabilities expand possibilities for next-generation biologics.
Photocleavable protecting groups and machine-learning-guided optimization of coupling conditions represent additional 2024–2026 developments. One notable 2025 clinical trial preparation study used AI-driven retrosynthesis planning to reduce development time for a novel antimicrobial peptide from 18 months to 4 months. Such tools are expected to accelerate translation from bench to bedside while maintaining the rigorous quality standards required by regulatory agencies.
Investigational techniques such as DNA-templated synthesis and solid-supported membrane synthesis remain pre-clinical as of April 2026 and are clearly distinguished from approved manufacturing routes.
Several blockbuster medications rely on robust peptide synthesis. Semaglutide, approved by the FDA for type 2 diabetes (2017) and weight management (2021), is produced via SPPS followed by solution-phase lipidation. Manufacturing data referenced in FDA review documents and subsequent 2022–2025 pharmacokinetic studies confirm >99% purity is routinely achieved at multi-kilogram scale.
Tirzepatide, a dual GLP-1/GIP receptor agonist approved in 2022, features a 39-amino-acid sequence with a C20 fatty diacid moiety. Synthesis involves Fmoc SPPS of the linear peptide followed by selective side-chain modification. Phase 3 trial publications (2022–2024) report consistent bioavailability across batches, attributing success to stringent control of aggregation-prone regions during synthesis.
Other FDA-approved examples include liraglutide (SPPS with palmitoylation), exenatide (synthetic version of exendin-4), and icatibant (a bradykinin B2 antagonist). A 2023 systematic review of 27 approved peptide APIs found that 78% are manufactured exclusively by chemical synthesis rather than recombinant expression, primarily due to the need for non-canonical amino acids or specific modifications incompatible with biological systems.
The FDA’s 2023 guidance on peptide drug substances emphasizes comparability protocols when scaling synthesis methods. Any process change—such as switching resins or coupling agents—requires demonstration of equivalent impurity profiles and biological activity. This regulatory framework ensures patient safety while encouraging continuous manufacturing improvements.
The following table summarizes key attributes based on 2020–2026 evidence:
| Method | Typical Length | Key Advantages | Main Limitations | Representative FDA-Approved Drugs | Average Crude Purity (2020–2025 data) |
|---|---|---|---|---|---|
| Solid-Phase (Fmoc) | 5–50 residues | Rapid, automated, scalable | Aggregation for >40 residues, solvent use | Semaglutide, liraglutide, icatibant | 75–92% |
| Liquid-Phase | 2–20 residues | High purity for short fragments, easier scale-up | Labor intensive, lower throughput | Exenatide (hybrid) | 85–95% |
| Recombinant Expression | >50 residues | Cost-effective for large peptides, aqueous | Limited to natural amino acids, host-cell impurities | Insulin (analogues), certain hormones | N/A (post-purification >98%) |
| Hybrid (SPPS + Ligation) | 30–100 residues | Combines precision with length | Multiple purification steps | Tirzepatide (partial) | 70–88% (pre-ligation) |
| Flow Chemistry SPPS | 5–70 residues | Real-time monitoring, reduced solvent | Higher equipment cost | Emerging APIs (investigational) | 88–96% |
Data compiled from meta-analyses and manufacturing case studies published 2021–2025. Hybrid approaches are increasingly favored for complex molecules requiring both non-natural modifications and extended sequences.

Despite advances, peptide synthesis presents technical and safety challenges. Common impurities include diastereomers from racemization and aspartimide formation, which can alter pharmacological activity. A 2024 meta-analysis linked certain synthetic impurities to injection-site reactions observed in early GLP-1 agonist formulations, prompting reformulation and tighter specifications.
Immunogenicity remains a concern for longer or heavily modified sequences. FDA guidance updated in 2022 requires assessment of anti-drug antibodies in all peptide therapeutics. Manufacturing facilities must comply with stringent cleanroom standards to prevent microbial contamination during cleavage and purification.
Environmental and worker safety issues associated with TFA, piperidine, and DMF have driven regulatory pressure toward greener processes. Publications from 2023–2026 document successful pilot-scale transitions to safer solvent systems without compromising product quality.
For off-label or investigational peptides synthesized in research laboratories, the FDA prohibits use in human subjects without an active Investigational New Drug (IND) application. All therapeutic claims require full clinical development pathways.
Looking toward the remainder of the decade, evidence points to broader adoption of artificial intelligence for sequence optimization and predictive impurity modeling. Continuous manufacturing platforms integrating synthesis, purification, and formulation are under active investigation in 2025–2026 pilot studies, promising reduced costs for chronic-use medications.
Multifunctional peptides combining targeting, therapeutic, and diagnostic properties (theranostics) represent an emerging area supported by early-phase trial data. Advances in orthogonal protection schemes and click chemistry are expected to facilitate site-specific conjugation at scale.
Sustainability will remain a priority, with regulatory incentives likely for processes demonstrating measurable reductions in process mass intensity. As new peptide modalities such as macrocyclic peptides and peptidomimetics advance through pipelines, synthesis methods must evolve in parallel to maintain the high purity and reproducibility demanded by FDA standards.
Ongoing collaboration between academic labs, contract development and manufacturing organizations (CDMOs), and regulatory bodies will be essential to translate these innovations into approved therapies that improve patient outcomes while minimizing environmental footprint.
Peptide synthesis stands as a mature yet rapidly evolving technology that underpins a growing segment of the pharmacotherapy landscape. From established solid-phase methods to cutting-edge flow chemistry and chemoenzymatic approaches, the evidence accumulated between 2020 and April 2026 demonstrates clear progress in yield, purity, scalability, and sustainability. These improvements have directly enabled the successful development and commercialization of transformative medicines such as semaglutide and tirzepatide, which have reshaped treatment paradigms for metabolic diseases.
The reviewed literature consistently emphasizes the necessity of rigorous analytical controls, regulatory compliance, and medical supervision when working with synthesized peptides. While FDA-approved products have robust safety profiles when manufactured under cGMP, investigational peptides synthesized outside approved pathways carry unknown risks and should not be used clinically. Researchers and developers are encouraged to consult the latest FDA guidances and peer-reviewed protocols when designing synthesis campaigns.
Future success will depend on balancing innovation with reliability—leveraging artificial intelligence, green chemistry, and hybrid methodologies while maintaining the stringent quality standards required for human therapeutics. As the pipeline of peptide-based drugs continues to expand, ongoing peer-reviewed evaluation and transparent reporting of synthesis methods will remain critical for advancing evidence-based pharmacotherapy.
Continued investment in scalable, sustainable synthesis technologies promises to make peptide drugs more accessible globally. Clinicians, scientists, and policymakers should monitor emerging data closely, always prioritizing patient safety and regulatory adherence in this dynamic field.
Word count: 2478

Peptide synthesis stands as a mature yet rapidly evolving technology that underpins a growing segment of the pharmacotherapy landscape. From established solid-phase methods to cutting-edge flow chemistry and chemoenzymatic approaches, the evidence accumulated between 2020 and April 2026 demonstrates clear progress in yield, purity, scalability, and sustainability. These improvements have directly enabled the successful development and commercialization of transformative medicines such as semaglutide and tirzepatide, which have reshaped treatment paradigms for metabolic diseases.
The reviewed literature consistently emphasizes the necessity of rigorous analytical controls, regulatory compliance, and medical supervision when working with synthesized peptides. While FDA-approved products have robust safety profiles when manufactured under cGMP, investigational peptides synthesized outside approved pathways carry unknown risks and should not be used clinically. Researchers and developers are encouraged to consult the latest FDA guidances and peer-reviewed protocols when designing synthesis campaigns.
Future success will depend on balancing innovation with reliability—leveraging artificial intelligence, green chemistry, and hybrid methodologies while maintaining the stringent quality standards required for human therapeutics. As the pipeline of peptide-based drugs continues to expand, ongoing peer-reviewed evaluation and transparent reporting of synthesis methods will remain critical for advancing evidence-based pharmacotherapy.
Continued investment in scalable, sustainable synthesis technologies promises to make peptide drugs more accessible globally. Clinicians, scientists, and policymakers should monitor emerging data closely, always prioritizing patient safety and regulatory adherence in this dynamic field.
Word count: 2478